
Gli oppiacei sono tra i migliori strumenti a disposizione del medico per il trattamento del dolore conseguente a danni tissutali e infiammazioni. Tuttavia, possiedono un lato oscuro di primaria importanza consistente in effetti collaterali da moderati a molto seri come sonnolenza, stitichezza, depressione respiratoria e elevata capacità di indurre dipendenza.
Un problema quest'ultimo particolarmente importante negli USA dove si parla apertamente di vera e propria epidemia legata all'abuso di sostanze antidolorifiche, prodotti di facile accesso grazie alle prescrizioni mediche (--> The Guardian).
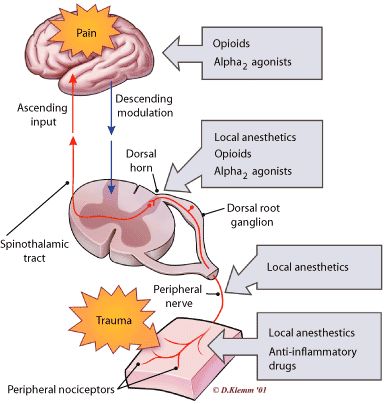
L'effetto analgesico degli oppiacei si attua su almeno tre livelli:
Un problema quest'ultimo particolarmente importante negli USA dove si parla apertamente di vera e propria epidemia legata all'abuso di sostanze antidolorifiche, prodotti di facile accesso grazie alle prescrizioni mediche (--> The Guardian).
L'effetto analgesico degli oppiacei si attua su almeno tre livelli:
- a livello spinale mediante il blocco presinaptico nella corno posteriore del midollo;
- nel tronco encefalico, con la stimolazione della sostanza grigia periacqueduttale che collegandosi al rafe magno spegne i neuroni nocicettivi specifici;
- modulazione della percezione del dolore.
 |
| Il controllo del dolore avviene a più livelli |
Fatta salva la necessità (soprattutto in senso etico) di neutralizzare per quanto possibile il dolore è ugualmente importante minimizzare gli effetti collaterali e il rischio di dipendenza (fortemente legato all'attivazione del circuito della ricompensa). Per questo fine una delle vie percorribili è la sintesi di farmaci in grado di funzionare solo dove necessario.
Facile a dirsi nel caso di lesioni periferiche o superficiali, molto meno quando il dolore è interno o oramai cronicizzato con tutto il carico di modificazioni neuronali che questo comporta. In questi casi gli analgesici "utili" sono di terzo livello (in genere oppiacei) i quali funzionano legandosi a recettori "profondi" nel sistema nervoso.
Nello specifico, il bersaglio funzionale sono i recettori oppioidi, il punto di azione naturale di molecole endogene come le encefaline e la beta-endorfina.
Tre sono le classi di recettori noti:
- μ: genera analgesia (a livello sovraspinale), miosi, depressione respiratoria, riduzione motilità gastrointestinale, euforia;
- k: genera analgesia (a livello spinale), miosi, depressione respiratoria, disforia (a differenza dei recettori μ), riduzione motilità gastrointestinale;
- δ: non genera analgesia, ma diminuisce il transito intestinale e deprime il sistema immunitario.
L'oppiaceo per antonomasia, la morfina, agisce come agonista sui recettori di classe μ e solo parzialmente su quelli di classe δ. Un farmaco ideale dovrebbe in qualche modo funzionare solo sui recettori μ (da qui in poi MOR) coinvolti nel circuito analgesico voluto evitando però ogni effetto su respirazione e mobilità intestinale. Ancora più importante, il farmaco non dovrebbe agire sulle cellule del circuito della ricompensa in modo da minimizzare i problemi di dipendenza.
Il problema è che trattandosi dello stesso recettore, sebbene localizzato su cellule o in distretti diversi, non è possibile che l'oppiaceo leghi solo il recettore sulle cellule del circuito X ma non del circuito Y; problema rafforzato dal fatto che la somministrazione del farmaco in questi casi avviene per endovena o con catetere spinale, rendendo quindi "automatica" la diffusione sistemica del farmaco e quindi l'attivazione generalizzata dei MOR.
Il team di Christoph Stein ha pubblicato nei giorni scorsi su Science i risultati di uno studio centrato proprio sullo sviluppo di farmaci analgesici ad azione mirata o meglio sulla modificazione di un farmaco già in uso in modo da renderlo funzionante solo nei tessuti danneggiati.
Punto di partenza è stato il considerare che lo stato infiammatorio associato al dolore - come nel caso di artrite, neuropatie o intervento chirurgico - è quasi sempre accompagnato all'acidificazione del tessuto colpito.
Con questa premessa i ricercatori si sono chiesti se fosse possibile sviluppare un farmaco agonista in grado di legare i recettori solo in condizioni di basso pH, lasciando così liberi (quindi non attivando i segnali a cascata) i MOR nei tessuti a pH normale.
La chimica ci insegna che "predizioni" del genere si ottengono dall'analisi della struttura della molecola e in particolare dal parametro pKa. Il pKa è la costante di dissociazione di una molecola che permette di predire lo stato (protonato o meno) della molecola al variare del pH. Modificando la molecola è così possibile ottenere un pKa "utile" allo scopo.
Se vogliamo che la sua specificità aumenti, sia ristretta cioè solo ai recettori localizzati nei tessuti infiammati, bisognerà modificare la molecola in modo che il pKa abbia valore tra 6 e 7 (un pKa inferiore a 6 limiterebbe la sua funzionalità solo ai tessuti troppo infiammati e/o con danno avanzato).
Per seguire questa idea i ricercatori hanno per prima cosa eseguito simulazioni al computer della molecola scoprendo che si poteva ottenere il pKa desiderato inserendo un atomo di fluoro al posto di un idrogeno in diverse posizioni.
 |
| Il fluoro "decisivo" nel fentanyl modificato (alias NFEPP) |
Passo successivo è stato testare il miglior candidato disponibile (che abbreviamo per semplicità con NFEPP) mediante test di funzionalità e tollerabilità in modelli animali.
Anche qui i risultati sono stati positivi:
- mentre la somministrazione endovenosa di fentanyl provocava analgesia generalizzata, il NFEPP (sempre per via endovenosa) produceva l'effetto analgesico ricercato solo nelle zone infiammate.
- Alte dosi di NFEPP non provocavano (a differenza del fentanyl) sedazione o depressione respiratoria.
- A riprova della specificità di azione, il trattamento con un antagonista classico come il naloxone, incapace di penetrare la barriera ematoencefalica, eliminava completamente l'azione analgesica del NFEPP ma solo parzialmente quella del fentanyl. Il dato è la migliore dimostrazione che il NFEPP funziona solo a livello periferico.
- Infine i test critici miranti a escludere il rischio di effetti indesiderati legati all'attivazione dei recettori centrali (nel circuito della ricompensa e quelli responsabili di sedazione, alterazione motoria e depressione respiratoria) o di quelli intestinali (causa di stipsi). Ancora una volta, mentre il fentanyl attivava il circuito della ricompensa (quindi la dipendenza) e alterava attività come locomozione e defecazione, il NFEPP non mostrava alcuno di questi effetti anche alle dosi più alte.
Nota. Un'altra molecola che ha sfruttato strategie di sviluppo indirizzate ad un effetto più controllato sui MOR è la oliceridina, oggi in fase di sperimentazione clinica.
Fonte
- A nontoxic pain killer designed by modeling of pathological receptor conformations.
V. Spahn et al. (2017) Science, 355 (6328) pp.966-969





Nessun commento:
Posta un commento