Nuovi farmaci e costi connessi (parte 3)
(Continua da ---> parte 2)
Da quanto scritto nelle due parti precedenti comincia, spero, ad emergere la complessità intrinseca del lungo percorso che porta alla approvazione di un farmaco. E si noti che ho volutamente tralasciato tutti gli aspetti prettamente scientifici legati al processo di scoperta, di validazione e infine di "idoneità" farmacologica. Per una trattazione un poco più specifica, ma senza eccessivi tecnicismi, potete leggere la pagina sulla progettazione dei farmaci sul sito della Treccani (-->qui).
Entriamo adesso nell'ultima parte di questo breve viaggio analizzando alcuni dati finora solo menzionati.
- Tra le false credenze vi è quella riferita all'ingente numero di nuovi farmaci annualmente immessi sul mercato. Niente di tutto questo! In Europa il numero di farmaci approvati nel 2013 dall'Agenzia Europea del Farmaco (EMA) è pari a 44 (vedi qui per maggiori dettagli). Il numero non cambia di molto se si va sul ricco, per introiti potenziali, mercato americano. Nei due grafici che seguono risulta abbastanza chiaramente che il numero medio delle molecole approvate negli ultimi 20 anni non è cambiato di molto
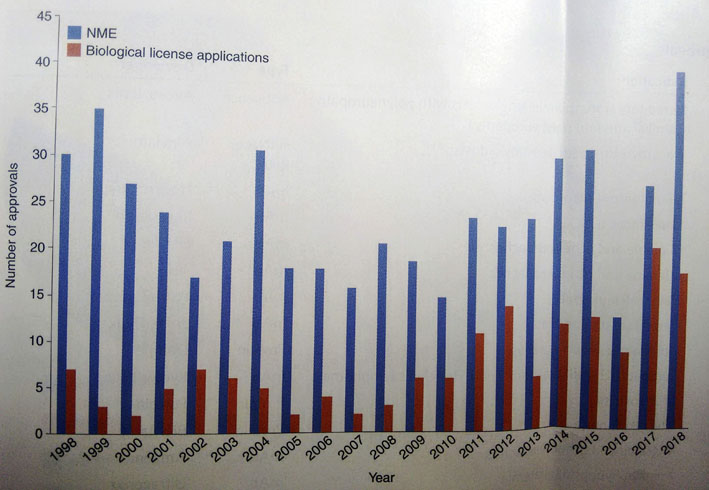 |
| Tabella aggiornata al 2018. Nuovi farmaci chimici (NME) o biologici (BLA) approvati negli ultimi 20 anni dalla FDA americana |
 |
| Un grafico simile del precedente (ma con un sottostante di molecole più ampio) in cui il confronto è centrato sull'aumento dei costi associati senza che vi sia stato un incremento dei prodotti. L'aumento delle conoscenze va infatti di pari passo con l'aumento dei controlli richiesti. |
 |
| Il ROI (Return on Investment) del settore Pharma negli ultimi 5 anni. Il valore si è più che dimezzato (credit: Nat. Rev. Drug Discov. 01/2016) |
- Vari sono i motivi che spiegano questi numeri e soprattutto il loro non aumentare, come invece il rapido avanzamento delle conoscenze biomediche avrebbe dovuto fare ipotizzare. Tra questi i sempre crescenti costi associati, dovuti paradossalmente alle maggiori conoscenze acquisite sulla complessità delle malattie, e l'aumento dei dati richiesti in sede di approvazione. Se infatti le conoscenze acquisite nell'ultimo decennio sono state incredibili anche le domande che le nuove conoscenze hanno generato (e che si traducono in controlli sempre più accurati) non sono meno rilevanti. Più si entra nel dettaglio dei meccanismi fisiologici e maggiori sono le cautele che l'ente regolatore impone. Il costantemente alto tasso di fallimento dei farmaci entrati nella sperimentazione clinica ha fatto il resto. Fallimento che non è dovuto a facilonerie nella progettazione dei farmaci ma proprio alla estrema difficoltà di creare molecole efficaci e sicure.
 |
| Nel grafico si vede chiaramente come i tempi necessari (e parliamo solo della fase finale, quella della valutazione) non sono cambianti negli ultimi 10 anni (dati riferiti a Europa, USA e Giappone). Se avessi voluto considerare i tempi per ottenere i dati necessari ad uno studio di fase 3, questi avrebbero potuto oscillare tranquillamente tra due e otto anni. |
- Altra domanda. Dove vanno a finire gran parte dei soldi spesi durante lo sviluppo di un farmaco.
 |
| Allocazione dei costi sostenuti per ogni fase dello sviluppo e validazione di un farmaco. Per intenderci, la sperimentazione animale risucchia circa 1/4 del costo totale ed è "sicura", nel senso che è uno dei pochi costi certi per l'industria. Indipendentemente dal fatto che il farmaco sia poi approvato (fine fase-III) o venga cancellato già in fase-I. Voi pensate veramente che se solo ne avesse la "libertà" una industria non cancellerebbe questi costi riversandoli in toto su altre metodiche di valutazione? |
Le ragioni principali del fallimento della sperimentazione (leggasi "non approvazione" o "interruzione della sperimentazione") di un nuovo farmaco sono riassunti nel grafico sottostante.
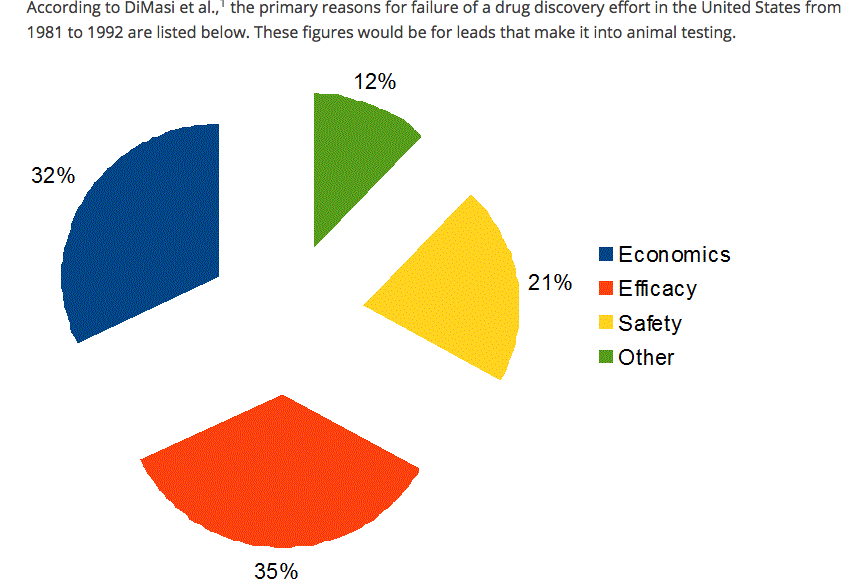
Particolarmente interessante è "entrare nel grafico" precedente per visualizzare alcuni dettagli molto significativi. Limitiamoci al decennio chiave 1991-2001 e scomponiamo le cause del fallimento di un farmaco (inteso come le cause che hanno portato alla non approvazione in sede regolatoria). Non solo ragioni cliniche ma anche economiche: scoprire che il farmaco sperimentale manca delle ipotizzate proprietà "superiori" rispetto al farmaco già esistente sul mercato lo rende un prodotto "inutile" per i "buyers" (servizio sanitario nazionale, assicurazioni, etc); altra causa importante di fallimento è scoprire che un farmaco "migliore" per le stesse indicazioni è entrato nella sperimentazione dopo il nostro per cui, anche se il proprio candidato ottenesse il semaforo verde, verrebbe poco dopo surclassato dal nuovo arrivato.
 |
| L'aumento delle conoscenze biochimiche ha permesso di minimizzare la farmacocinetica come causa di fallimento. Al contrario sono la componente commerciale è divenuta centrale (tipico esempio la presenza di un farmaco preesistente sul mercato e l'assenza di migliorie sostanziali in efficacie o sicurezza del nuovo farmaco) |
- La figura successiva vuole invece evidenziare come varia la percentuale di successo nel passaggio alla fase successiva durante le diverse tappe della sperimentazione clinica.
 |
| Apparentemente superare la fase 3 è più facile che superare la fase 2. In realtà il dato deve essere pesato per il numero di sperimentazioni che pur avendo avuto successo in fase 2, non proseguono oltre per motivi vari (presenza di un concorrente di uguale affidabilità sul mercato, rischio di insuccesso elevato, brevetto in fase di scadenza, ...). Un aggiornamento a questi valori viene da uno studio pubblicato nel 2016. Le percentuali di successo da fase 1 a registrazione sono (come media) pari a 63%, 31%, 58% e 85%; il che equivale ad 9,6% complessivo. Tale valore cambia a seconda sia del campo di applicazione della molecola con percentuali fino al 26% in ematologia e 5% in oncologia. |
- Da un punto di vista delle risorse investite è sempre meglio che un prodotto "fallisca" farmacologicamente PRIMA che DOPO. Un fallimento in fase 1 (sicurezza e tollerabilità) o fase 2 (efficacia e sicurezza) riguarda studi clinici con una decina o qualche centinaio di soggetti analizati. Un fallimento in fase 3 (ad esempio il farmaco funziona ma non così bene come atteso o non meglio di quello che esiste sul mercato) sarebbe successivo a trial clinici superiori ai 3 anni e con migliaia di soggetti analizzati per un costo sottostante anche di centinaia di milioni di euro.

Criteri di selezione molto stringenti in fase 2 rendono più facile il successo in fase 3 dei farmaci "sopravvissuti". Un esempio tipico nel grafico è quello dei farmaci respiratori ("resp"). La selezione che falcidia il 90% dei candidati in fase iniziale fa si che il 90% di quelli entrati nella costosa e lunga fase 3 abbia successo. Non sempre è possibile ("Neuro-D") ma è una strategia intelligente (credit: UCSD) - Va bene. Tutto chiaro. Ma manca ancora una informazione fondamentale. A quanto ammontano nel concreto i costi? Reggetevi forte. Il costo stimato per farmaco approvato è di poco superiore a 1 miliardo di dollari! Avete letto bene. Un miliardo di dollari (nota. Il dato più aggiornato riferito al 2015 indica in 1,51 miliardi di dollari il valore medio) E' ovvio che queste stime risentano dei diversi metodi di calcolo e del fatto che si tratta di medie. Se uno infatti prendesse come riferimento il caso limite dei costi sostenuti da una azienda appena nata che ha comprato per nulla il brevetto sviluppato da un'altra azienda fallita, evitando così di investire in ricerca e sviluppo e che ha avuto la fortuna di passare indenne e abbastanza velocemente attraverso tutte le fasi della preclinica e della clinica, allora il costo reale sostenuto da questa azienda fortunata sarebbe ben inferiore ma pur sempre intorno a qualche centinaio di milioni di dollari. E siamo appunto in un caso limite in cui di fatto i costi strutturali e di sviluppo non esistono. Sarebbe come calcolare i tempi di percorrenza autostradali casa-lavoro (e il consumo di benzina) nella settimana di ferragosto e generalizzarli per tutto il resto dell'anno. I costi indiretti sono molto elevati e nell'esempio precedente non tenevano conto della liquidità bruciata dalla azienda fallita. Nella realtà bisogna quindi tenere conto dei costi sostenuti dall'azienda per sviluppare e testare tutte le molecole che hanno preceduto quella che e' riuscita ad arrivare sul mercato. Tanto più sono le molecole che si sono perse per strada in fase avanzata, tanto maggiore sarà l'ammontare dei costi consolidati.

Riassumendo brevemente, tra le voci di spesa abbiamo la ricerca di base, i test preclinici (ivi compresi gli ingenti costi dei test sugli animali) e infine la lunga (a volte imprevedibilmente lunga) fase di validazione sugli umani che iniziano con il profilo di sicurezza e dosaggio sui volontari sani e si concludono sulla validazione dell'efficacia e della sicurezza su migliaia di pazienti. E ricordiamoci dei costi legati al marketing senza il quale ben difficilmente un medico potrebbe venire a scoprire dell'esistenza di un prodotto migliore per un suo paziente.
Un dato emerge chiaramente, troppo spesso trascurato da chi contesta ferocemente la sperimentazione animale e attribuisce alla lobby farmaceutica il desiderio di continuarla nonostante sia, a loro avviso, inutile o sostituibile con mezzi più semplici. E' il caso di ricordare che, giusto per essere cinici, il costo derivante dalla sperimentazione animale, tra gestione stabulari e test, è un costo non indifferente che qualunque azienda eliminerebbe immediatamente se non fosse obbligata a fornire i risultati dei test associati. Non ho mai sentito di aziende che tagliano a destra e a manca per risparmiare sui costi ma che tengono in piedi a loro spese un distruttore di liquidità monetaria come gli stabulari, se non fosse strettamente necessario.
La stima reale dei costi sostenuti in ciascuna fase della sperimentazione clinica è complicata dalle metodologie di calcolo usate. Si va da stime di 43M $ per uno studio facile a 2,9 miliardi di $ "buttati" complessivamente per un farmaco ritirato dopo essere stato approvato- Nella versione completa si computano costi come il "costo-opportunità" (quanto mi avrebbe reso se invece di investire i soldi in uno studio poi fallito li avessi usati per portare avanti una molecola con maggiori chance di successo e mercato?), il costo di produzione e la tipologia di studio usato. Uno studio recente ha preso in esame i costi sostenuti per i 59 farmaci approvati dalla FDA nel biennio 2015-16 e si va da 2M $ per uno studio di fase 3 con soli 4 pazienti (patologia molto rara) ai 347M $ per uno studio di non inferiorità di un farmaco ad azione cardiovascolare che ha coinvolto 8 mila pazienti. Costi del solo studio clinico, senza costare le spese di produzione e costo-opportunità. Uno studio simile condotto poco prima e centrato su 700 studi clinici condotti dalle 7 principali pharma ha fornito valori medi per la sola parte clinica di 3,4M $ (fase 1), 8,6M $ (fase 2) e 21,4M $ (fase 3).
 |
| Schema riassuntivo dei costi associati allo sviluppo di un un singolo farmaco. Per ogni farmaco che non viene approvato o che si perde in uno dei passaggi intermedi, il costo per l'azienda è duplice: da una parte il costo reale di poche decine di milioni di dollari quando va bene (cioè quando la molecola si rivela non adatta e/o tossica nelle primissime fasi); dall'altra i cosiddetto costo-opportunità cioè l'avere perso soldi che potevano essere usati per malattie meno "etiche" ma di sicuro successo commerciale (ad es. un clone del Viagra privo di effetti collaterali) |
- Il prezzo di vendita. Altro aspetto poco conosciuto è quello a cui ho accennato brevemente nella seconda parte, cioè che all'approvazione del farmaco non segue in automatico l'entrata in commercio. Nella maggior parte dei paesi questo passaggio richiede un ulteriore valutazione. Il farmaco, approvato da un punto vista clinico, deve vedersela con il giudizio dei pagatori sul fatto che sia o meno indispensabile. Chi sono i pagatori? Non i pazienti ma chi di fatto paga la gran parte del prezzo di vendita reale del farmaco, cioè il servizio sanitario nazionale nella vece di AIFA (o equivalenti in altri paesi) e le assicurazioni (specialmente in USA). Questi sono gli interlocutori con cui una azienda farmaceutica deve interfacciarsi per stabilire il prezzo di un farmaco. Si parte sempre da un concetto base: tutti questi pagatori hanno un certo budget che per quanto grande sia NON è infinito. I soldi devono essere in grado di coprire il fabbisogno farmaceutico di tutta la popolazione, sia per i farmaci di uso corrente che per quelli usati da un ridottissimo numero di persone e che quindi costano molto di più. Ogni nuovo farmaco che entra attinge a questo fondo e quindi diminuisce la copertura complessiva. Questo il motivo per cui in periodi di crisi compaiono i famigerati ticket; l'unico modo per rimpinguare la cassa senza diminuire la copertura effettiva. Il confronto tra l'azienda e i pagatori è sul prezzo. L'azienda vuole ottenere un prezzo di vendita più alto possibile (almeno in grado di coprire le spese) mentre i pagatori, che di questo prezzo pagheranno gran parte se non tutto, si opporranno. In alcuni casi si arriva perfino all'accordo che una azienda farmaceutica ritira un farmaco "vecchio" in cambio dell'accettazione di uno nuovo (leggi anche QUI per approfondimenti). Attenzione non ci sono qui "cattivi" e "buoni" ma due legittimi gruppi di interesse. In questo ambito l’Agenzia Italiana del Farmaco (AIFA) è l’autorità nazionale competente per l’attività regolatoria dei farmaci in Italia che rilascia il permesso di entrata sul mercato italiano.
Alla fine di questa lunga, ma allo stesso tempo semplificata, cavalcata sul processo che porta alla approvazione di un farmaco spero risulti chiara l'importanza di conoscerne gli aspetti meno noti.
Ignorare questi passaggi chiave spiega come sia stato possibile da parte
di molte persone prendere in considerazione una non-terapia come
Stamina o prima la terapia Di Bella. Trattamenti privi non solo di dati concreti riferiti a efficacia e sicurezza ma nemmeno mai sottoposti ai processi di validazione e
controllo rigoroso che solo i protocolli sperimentali permettono di ottenere.
Un altro modo per ottimizzare gli investimenti fatti nello sviluppo di un farmaco è quello di trovarne utilizzi per indicazioni non previste inizialmente. Di seguito una tabella che illustra i casi più famosi di farmaci i cui effetti inattesi (in alcuni casi veri e propri effetti collaterali non voluti e/o gravi - vedi il caso Viagra e Talidomide ) si sono rivelati fondamentali per il trattamento di patologie del tutto diverse da quelle originariamente pensate
 |
| Altri esempi di farmaci in uso o dismessi per cui è stata trova una nuova "vita" li trovate seguendo il tag --> NewOldDrugs |
Tutto questo spiega i prezzi ... almeno fino a quando non si attivano accordi dietro le quinte che provocano aumenti generalizzati anche per i farmaci "obsoleti". Un problema tipico per la sistema sanitario americano basato sulle assicurazioni, in cui il "pricing" varia a seconda del potere di ottenere sconti (o dalla convenienza a chiederli) da parte dell'assicurazione. Problema trattato nell'articolo --> "Shadow pricing and the art of profiteering from outdated therapies".
(FINE)
Per volesse approfondire l'argomento posso consigliare alcuni link come base di partenza:
- Il ruolo di AIFA nella ricerca clinica (pdf)
- Agenzia Italiana del Farmaco (AIFA) --> "Sperimentazione clinica" e "come nasce un farmaco".










.jpg/600px-Hippocampus_(brain).jpg)